- Theoretical Designs of Advanced Battery Materials
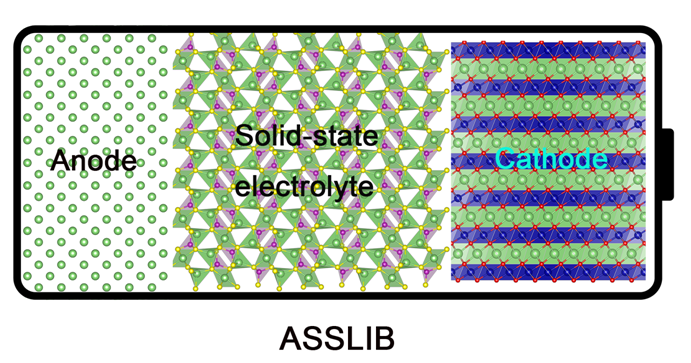
All-solid-state lithium-ion batteries (ASSLIBs) with high energy density are promising to substitute the liquid organic electrolyte based batteries, which enable more compact stacking structure and increased safety of next-generation high-energy-density batteries and power devices, e.g., electric vehicles. However, the interfacial compatibility between electrodes and electrolyte is still one of the biggest challenges for applying solid electrolytes in ASSLIBs. Therefore, we not only devote ourselves to screening superionic conductors for ASSLIBs based on DFT calculations, but also understanding the interfacial transport mechanism at atomic and electronic levels for the advanced design of the compatible interfaces for the next-generation ASSLIBs.
- Yang, Y.; Guan, C.; Ouyang, R.; Zhu, H. Harvest the Polyanion Rotation in Sodium Superionic Conductors? Chemistry of Materials 2024, 36 (8), 3776–3785. pdf
- Guan, C.; Yang, Y.; Ouyang, R.; Jing, H.; Yan, J.; Li, G.; Duan, H.; Zhu, H. Unlocking the Chemical Space in Anti-Perovskite Conductors by Incorporating Anion Rotation Dynamics. Energy Storage Materials 2023, 62, 102936. pdf
- Ouyang, R.; Xu, Z.; Zhu, H. Correlated Factors for Li-Ion Migration in Ionic Conductors with the Fcc Anion Sublattice. The Journal of Chemical Physics 2023, 158 (17), 174705. pdf
- Xu, Z.; Chen, X.; Chen, R.; Li, X.; Zhu, H. Anion Charge and Lattice Volume Dependent Lithium Ion Migration in Compounds with Fcc Anion Sublattices. npj Computational Materials 2020, 6 (1), 1–8. pdf
- Xu, Z.; Zhu, H. Anion Charge and Lattice Volume Maps for Searching Lithium Superionic Conductors. Chemistry of Materials 2020, 32 (11), 4618–4626. pdf
-
Mg Alloy Design With Good Corrosion Resistance And Mechanical strength
As the lightest structural materials with high specific strength, Mg has the potential to be used in automobile industries and aerospace fields. However, the poor corrosion and ductility of Mg alloys limit their wide applications. In this project, we focus on two aspects: (1) determining the polarization curves for Mg alloys and their thermodynamic stability in aqueous environment based on DFT simulations and experimental data to understand different alloying elements’ effects; (2) studying the structure, electronic property and mechanical strength of Mg alloy matrix and precipitations. We are also interested in the properties of other important interfaces, for example, the Ni-Ni3Al interface design through appropriate alloying.
- Wang, Y.; Tang, Q.; Xu, X.; Weng, P.; Ying, T.; Yang, Y.; Zeng, X.; Zhu, H. Accelerated Discovery of Magnesium Intermetallic Compounds with Sluggish Corrosion Cathodic Reactions through Active Learning and DFT Calculations. Acta Materialia 2023, 255, 119063. pdf
- Wang, Y.; Xie, T.; Tang, Q.; Wang, M.; Ying, T.; Zhu, H.; Zeng, X. High-Throughput Calculations Combining Machine Learning to Investigate the Corrosion Properties of Binary Mg Alloys. Journal of Magnesium and Alloys 2022, 12 (4), 1406–1418. pdf
- Z. Luo, H. Zhu, T. Ying, D. Li, X. Zeng, “First-principles calculations on the influence of solute elements and chlorine adsorption on the anodic corrosion behavior of Mg (0001) surface”, Surface Science 2018. pdf
-
Thermoelectric and Piezoelectric Materials Design
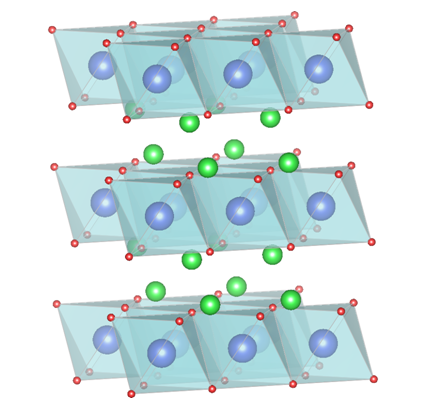
Thermoelectric (TE) materials with the ability to convert temperature differences to electric voltage and high chemical stability can be used in power generations from waste heat or in extreme conditions such as outer space. Recently, a group of phonon glass-electron crystal (PGEC) with layered structure has been reported with ultra-low thermal conductivity and decent power factor. However, the discovery and improvement of this group of materials is limited by the long and costly process to synthesize, optimize and measure thermoelectric properties. Here, we perform a comprehensive study on the stability and thermoelectric performance of thousands of layered TE materials, and uncover the design principles for promising PGEC TE materials, based on high throughout density functional theory (DFT) calculations and machine learning.
- Jing, H.; Guan, C.; Yang, Y.; Zhu, H. Machine Learning-Assisted Design of AlN-Based High-Performance Piezoelectric Materials. Journal of Materials Chemistry A 2023, 11, 14840. pdf
- Gibbs, Z. M.; Ricci, F.; Li, G.; Zhu, H.; Persson, K.; Ceder, G.; Hautier, G.; Jain, A.; Snyder, G. J., Effective mass and Fermi surface complexity factor from ab initio band structure calculations. npj Computational Materials 2017, 3, (1), 8. pdf
- Aydemir, U.; Pöhls, J. H.; Zhu, H.; Hautier, G.; Bajaj, S.; Gibbs, Z. M.; Chen, W.; Li, G.; Ohno, S.; Broberg, D., YCuTe2: a member of a new class of thermoelectric materials with CuTe4-based layered structure. Journal of Materials Chemistry A 2016, 4, (7), 2461-2472. pdf
- Zhu, H.; Hautier, G.; Aydemir, U.; Gibbs, Z. M.; Li, G.; Bajaj, S.; Pöhls, J. H.; Broberg, D.; Chen, W.; Jain, A., Computational and experimental investigation of TmAgTe2 and XYZ2 compounds, a new group of thermoelectric materials identified by first-principles high-throughput screening. Journal of Materials Chemistry C 2015, 3, (40), 10554-10565. pdf
-
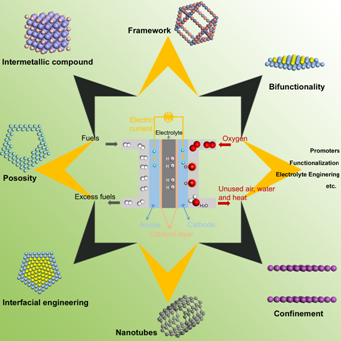
New Design Principles for Advanced Catalysts
As a promising avenue for small to intermediate scale operations, fuel cell devices play a vital role in energy generation and utilization technologies. However, the slow kinetics of oxygen reduction reaction (ORR) as the critical step of electrochemical oxidation of a fuel (typically hydrogen, methanol, etc.) limits the development of fuel cell devices. The classical volcano relationship on the basis of the two- and four-electron oxygen reduction pathways proposed by Nørskov demonstrates limited success in bringing down the ORR overpotential. Thus, we are especially interested in finding a new strategy to break the traditional volcano relationship and design catalysts superior to platinum by combing high-throughput simulations, materials physics, chemistry, and experiment.
- Qin, Y.; Li, F.; Tu, P.; Ma, Y. L.; Chen, W. L.; Shi, F. L.; Xiang, Q.; Shan, H.; Zhang, L. F.; Tao, P.; Song, C. Y.; Shang, W.; Deng, T.; Zhu, H.; Wu, J. B., Ag3PO4 electrocatalyst for oxygen reduction reaction: enhancement from positive charge. RSC Advances 2018, 8 (10), 5382-5387. pdf
- Ma, Y.; Li, F.; Ren, X.; Chen, W.; Li, C.; Tao, P.; Song, C.; Shang, W.; Huang, R.; Lv, B.; Zhu, H.; Deng, T.; Wu, J., Facets Matching of Platinum and Ferric Oxide in Highly Efficient Catalyst Design for Low-Temperature CO Oxidation. ACS Applied Materials & Interfaces 2018. pdf
- Liu, Y.; Gu, X.; Qi, W.; Zhu, H.; Shan, H.; Chen, W.; Tao, P.; Song, C.; Shang, W.; Deng, T.; Wu, J., Enhancing the Photocatalytic Hydrogen Evolution Performance of a Metal/Semiconductor Catalyst through Modulation of the Schottky Barrier Height by Controlling the Orientation of the Interface. ACS Applied Materials & Interfaces 2017, 9, (14), 12494-12500. pdf
-
Improving The Stability of Perovskite Solar Cells
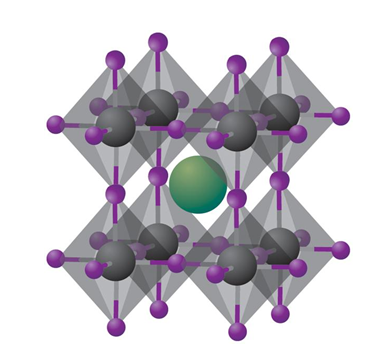
Hybrid organic-inorganic Perovskite is recently thought of as the 3rd new generation solar cell for its competitive power conversion efficiency and low-cost fabrication, while the instability is one of the main obstacles to being widely used in the photovoltaic field. Thus, we focus on improving the intrinsic stability of perovskite by chemistry and structural modification and understanding the correlation between phase stability and intrinsic atomistic level features. We also collaborate with the leading experimental group at SJTU.
- Wu, J.; Qi, W.; Luo, Z.; Liu, K.; Zhu, H., Electronic Structure and Stability of Lead-free Hybrid Halide Perovskites: A Density Functional Theory Study. Journal of Shanghai Jiaotong University (Science) 2018, 23, (1), 202-208. pdf
-
High-throughput Simulations on Generalized Stacking Fault Energies
The dislocation core structures and mobilities are partly controlled by the ease to shear a crystal along a given crystallographic plane, which can be characterized by the generalized stacking fault energy (GSFE). Thus, the determination of GSFE appears to be an instructive first step towards the understanding of the plasticity of materials. However, the success of such complex GSFE calculations usually relied heavily on the human intuition and manual operations on the atomic model building, simulation parameter adjustments, and final results analysis, which make it difficult for large-scale investigation of GSFEs of a variety of metals. Therefore, we focus on developing a high-throughput tool for calculating GSFE on typical slip systems of HCP, FCC and BCC structure metals based on density functional theory (DFT) simulations, and applying it to create the GSFE database for the elemental Materials.
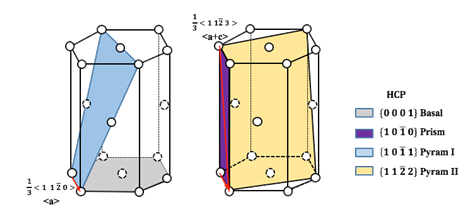
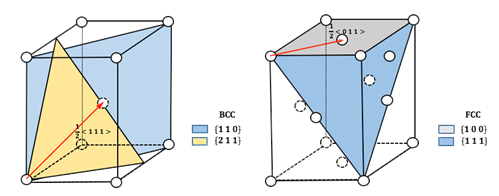
- Dong, Q.; Luo, Z.; Zhu, H.; Wang, L.; Ying, T.; Jin, Z.; Li, D.; Ding, W.; Zeng, X., Basal-plane stacking-fault energies of Mg alloys: A first-principles study of metallic alloying effects. Journal of Materials Science & Technology 2018. pdf