Research Goal
The main goal of our research is to establish an integrated, intelligent and insightful approach for accelerating new materials design and optimization, by advancing in the areas of multi-scale modeling, multi-physics modeling and machine learning.
Computational Methods
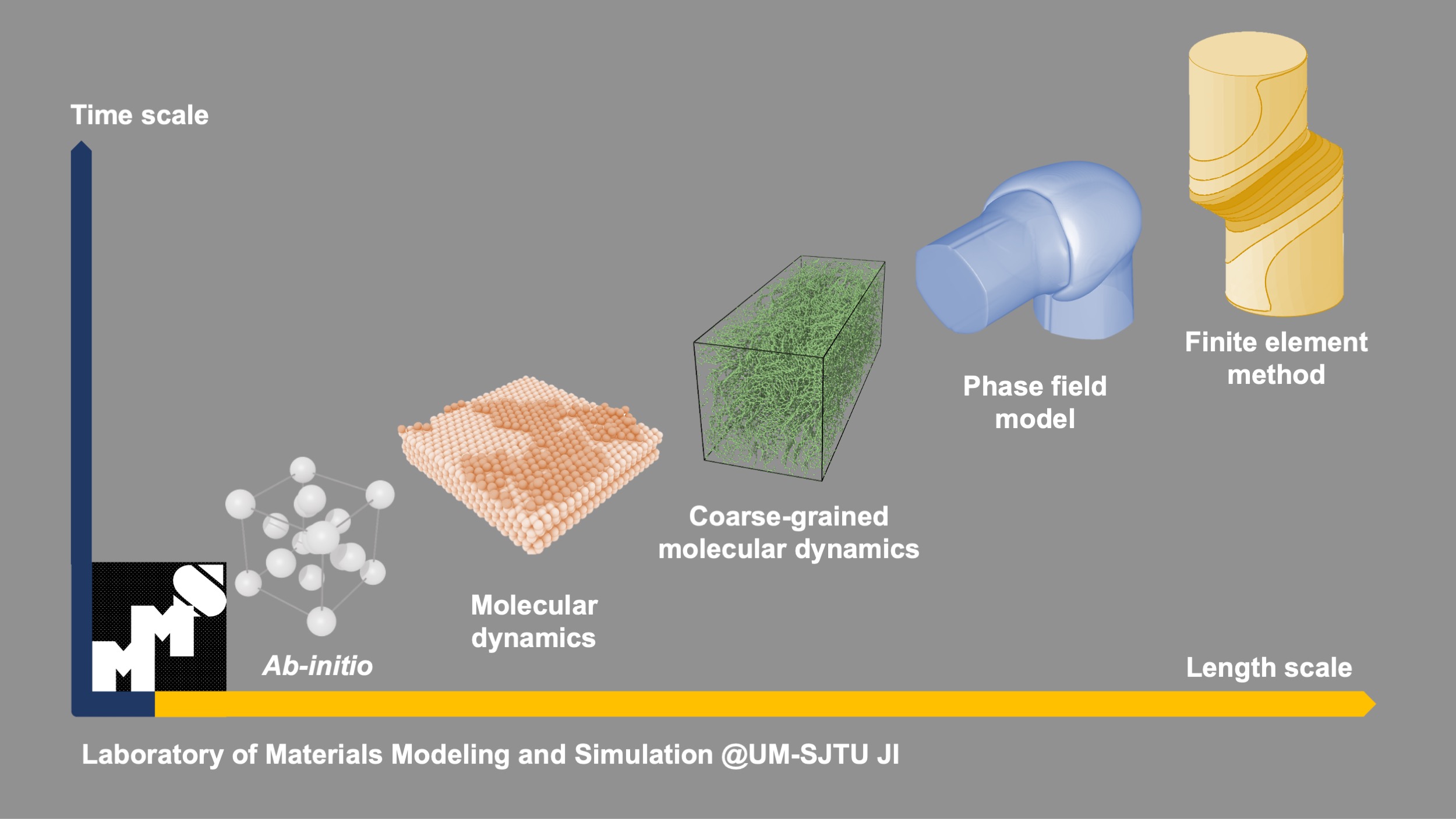
Our research involves a very wide range of materials modeling and simulation methods, from atomistic to continuum scale, including density functional theory, molecular dynamics, coarse-grained molecular dynamics, phase field model, finite element method and etc. The cooperation of computational methods at different scales enables accurate and systematic predictions on a variety of materials properties (such as electronic, mechanical, transport, interfacial and structural properties). In addition, this allows investigation of material synthesizability and stability, to promote the realization of promising new materials.
Material Systems
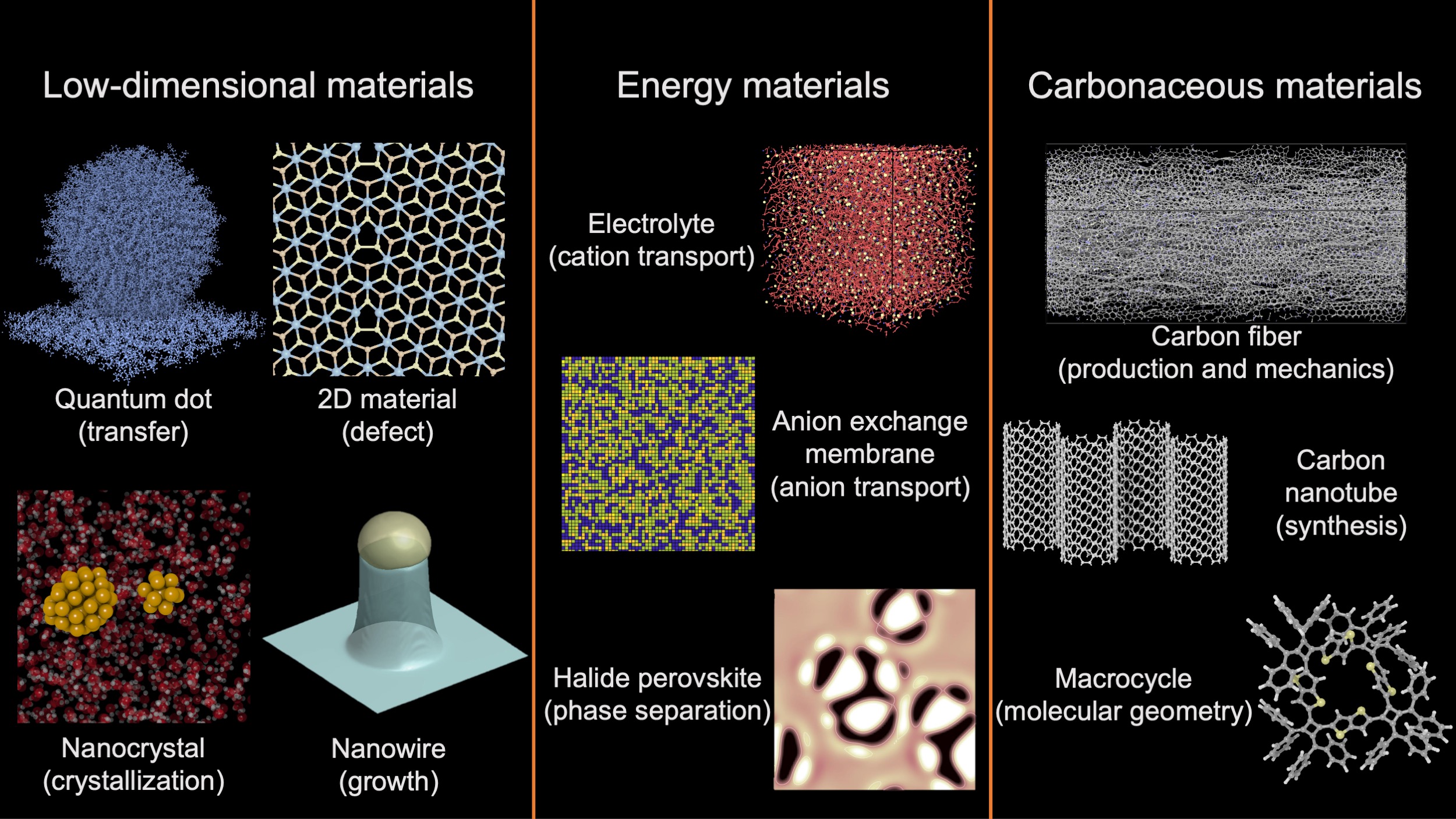
We list several representative material systems we have studied or we are currently working on, which can generally be categorized into the classes of low-dimensional materials, energy materials and carbonaceous materials.
Examples
Here we would like to briefly show a few examples from our recent work, and many of them were done in close collaboration with other research groups. Certainly they did not cover the entire spectrum of our research. If you would like to know more, please check our latest publication list or simply send us an email.
Polymer electrolyte design
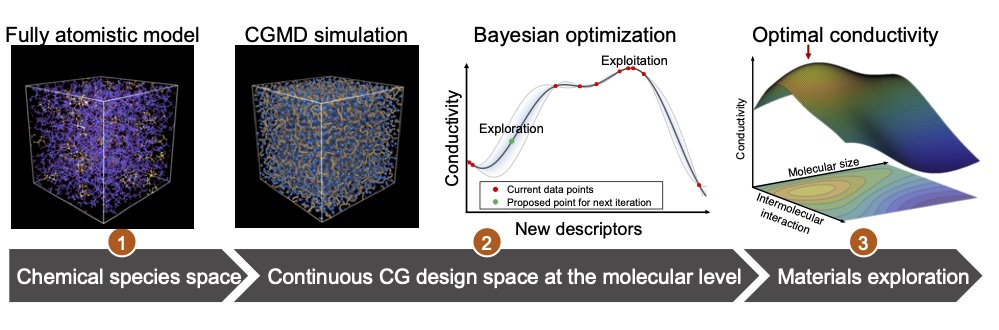
Here we aimed to accelerate the design of highly conductive polymer electrolyte materials for all-solid-state battery applications. Specifically, we first converted the conventional chemical species space to a continuous design space via the coarse-graining approach. Then we adopted the Bayesian optimization method for an efficient screening, to identify the collective effects of molecular level material properties on the ionic conductivity, based on which we would be able to achieve the global optimization of the system conductivity.
Nanowire structural change
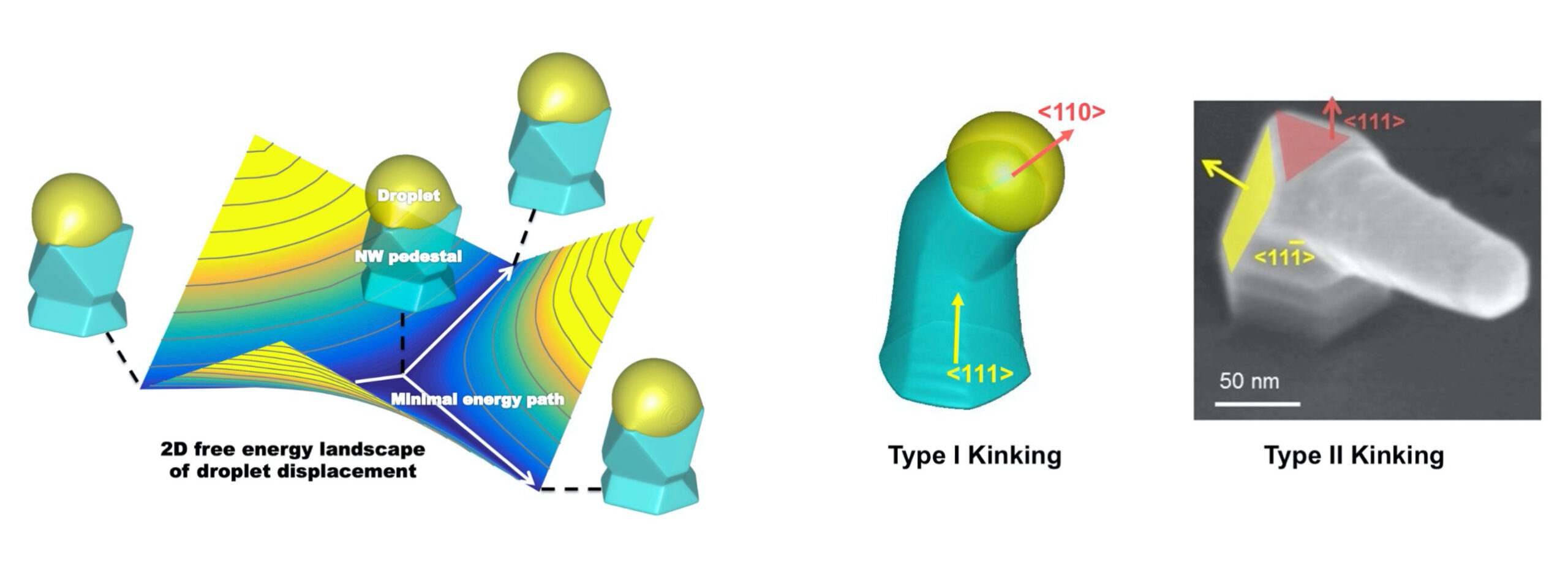
Here we developed a 3D multi-phase field model, which is capable of capturing the dynamical process of nanowire vapor-liquid-solid growth as well as reproducing the experimental wire geometries. With this model, we clarified the formation mechanisms of several abnormal semiconductor nanowire structures, and predicted the conditions at the onset of these structural changes.
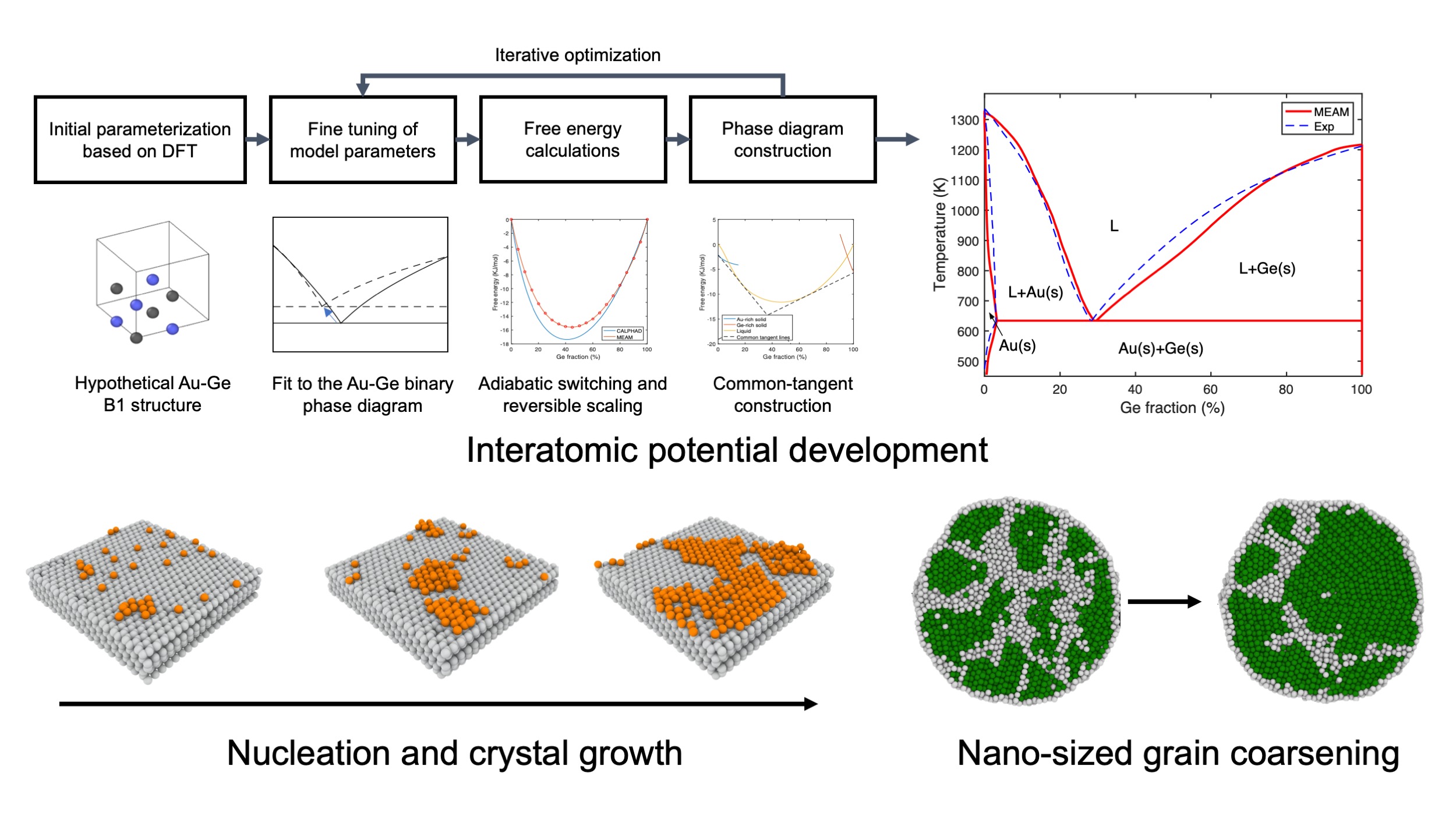
Crystallization mechanisms
Here we demonstrated our ability of developing an empirical interatomic potential well fitted to the binary phase diagram. With an accurate potential, we can perform molecular dynamics simulations to reveal the atomistic mechanisms of many interesting phenomena. For example, from the simulation trajectories, we can identify the interface morphology of gold-catalyzed silicon epitaxial growth; or we can capture the grain coarsening process inside a polycrystalline nanoparticle.